(西南大学 化学化工学院,重庆 400715)
摘要:基于密度泛函理论的第一性原理计算,我们研究了Sb(101)、Sb(110)、Bi(101)、Bi(110)四种晶面上CO2RR生成甲酸的反应机理,并探索了晶面相关的催化剂活性和产物选择性。
关键词:电催化;二氧化碳还原反应;甲酸
1.引言
为了缓解大气中二氧化碳(CO2)的过量积累及其引发的环境问题,迫切需要开发高效的CO2转化技术[1-2]。在温和的反应条件下,利用可持续电力进行电催化二氧化碳还原反应(CO2RR)是生产增值化学品的可行途径之一。电催化CO2RR技术可以实现碳减排和碳资源的循环利用,具有重要的环境效益[3]。其中,CO2转化为甲酸被认为是最有前景的反应之一。该反应只需要转移两个电子就可以生成一个甲酸分子,反应路径简单。此外,甲酸在常温常压下是液体,比H2更容易运输和储存。甲酸除可用作抗菌剂和燃料之外,还可以通过分解成CO2和氢气来储存高价值的H2,这些氢气可以循环利用,并通过CO₂RR再次生成甲酸。因此,甲酸可作为化学能量存储的平台[4-5]。
甲酸的形成与关键中间体*HCOO的生成密切相关。在催化剂表面,CO2通过吸附活化发生质子耦合电子转移反应,生成*COOH和*HCOO中间体(*表示吸附在催化剂表面)。*HCOO中间体是通过氧原子与催化剂表面结合,随后再进行一步质子电子耦合反应生成甲酸。质子可以是表面吸附的氢(*H)或者溶液中溶解的氢(*H),电子来源于电极内部。*COOH中间体通过碳原子与催化剂表面结合,在随后的质子耦合电子转移的反应中发生C-O键的断裂,生成CO和H2O。
由于CO2分子惰性较强,CO2还原反应动力学缓慢,需要较大的能量来激活C=O双键。此外,CO2还原为CO、析氢反应(HER)和甲酸的过程会相互竞争,导致产物选择性问题。因此,开发高效电催化剂以促进CO2选择性转化成甲酸显得尤为重要。
在这项工作中,我们研究了Sb基和Bi基催化剂上CO2RR的反应途径。我们选取了两种金属的(101)和(110)四个不同表面进行研究,结合电化学反应速率理论和第一性原理DFT模拟,建立了微观动力学模型,旨在深入探究两种p区金属的反应动力学和催化活性。通过结构优化,确定了热力学上最稳定的吸附构型,并研究了电极电位对反应速率和反应势垒的影响,揭示了热力学控制和动力学控制的反应路径之间的竞争关系。
2.计算细节
本文全部使用VASP软件进行电子结构计算。截断能为400 eV的平面波基组来表示价电子。采用投影子缀加波方法(PAW)来描述核电子相互作用。广义梯度近似(GGA)下的PBE泛函用于计算体系的交换相关能。电子自洽迭代的收敛标准为1.0![]() 10−5 eV,自由原子的最小力场设为0.02 eV/Å。采用隐式溶剂化模型VASPsol描述静电和分散对溶质-溶剂相互作用的影响,水的介电常数为78.4。在Materials Project晶胞库中选取Sb和Bi晶胞,两种晶胞均为R心六方晶胞,对其晶格常数进行优化,计算出平衡稳定状态下的晶格常数,Sb = 4.37 Å, Bi = 4.58 Å。对Sb(101)、Sb(110)、Bi(101)、Bi(110)晶胞采取底部两层固定。采用231的k点来进行几何优化。在垂直平面的z方向上,设置厚度为15 Å的真空层分隔晶胞中周期性重复的原子,以避免相邻金属原子层与层之间的镜像作用。
10−5 eV,自由原子的最小力场设为0.02 eV/Å。采用隐式溶剂化模型VASPsol描述静电和分散对溶质-溶剂相互作用的影响,水的介电常数为78.4。在Materials Project晶胞库中选取Sb和Bi晶胞,两种晶胞均为R心六方晶胞,对其晶格常数进行优化,计算出平衡稳定状态下的晶格常数,Sb = 4.37 Å, Bi = 4.58 Å。对Sb(101)、Sb(110)、Bi(101)、Bi(110)晶胞采取底部两层固定。采用231的k点来进行几何优化。在垂直平面的z方向上,设置厚度为15 Å的真空层分隔晶胞中周期性重复的原子,以避免相邻金属原子层与层之间的镜像作用。
小分子吸附到金属表面上,可以利用表达式(1)计算吸附小分子的结合能
![]() (1)
(1)
E*M,表面吸附分子的总能量;E*,洁净表面的能量;EM,气相中小分子的能量。E*M的值越小,越难在表面上脱附。
对于一个电极反应,根据表达式(2)计算出标准反应条件下(pH = 0,温度为298.15K,压力为1 atm)0 V vs SHE时的吉布斯自由能:
![]() (2)
(2)
EDFT是最稳定吸附构型的能量,EZPE,振动频率经过修正后得到的零点能,![]() Cp dT,是焓校正,TS是熵贡献。
Cp dT,是焓校正,TS是熵贡献。
CO2RR中任意一个单电子-质子转移反应式:
![]() (3)
(3)
表达式(3)的吉布斯自由能变ΔG = G*AH − G*A – GH+−Ge-,取决于溶液的pH和电极电势。在298.15K,pH = 0,电极电位为0 V时,反应H+ + e- → 1/2H2的ΔG = 0,即GH+ + Ge- = 1/2GH2。表达式(3)的反应自由能可以改写为ΔG = G*AH − G*A – 1/2 GH2。
本文采用Marcus微观理论动力学模型计算CO2还原的活化能:
 (4)
(4)
其中,ΔG是吉布斯自由能变,![]() R和
R和![]() P表示反应物和产物的总重组能。
P表示反应物和产物的总重组能。
3.结果与讨论
表1.U = 0 V vs.SHE时四个金属表面上反应的自由能和活化能(单位:eV)
反应类型 | Sb(101) | Sb(110) | Bi(101) | Bi(110) | |||||
G | Ea | G | Ea | G | Ea | G | Ea | ||
*CO2 + H+ + e-→*COOH | 0.80 | 1.28 | 0.65 | 1.21 | 0.77 | 1.14 | 0.68 | 1.19 | |
*CO2 + H+ + e-→*HCOO | 0.35 | 1.17 | 0.18 | 1.07 | 0.22 | 0.98 | 0.33 | 1.08 | |
*COOH + H+ + e-→*CO + *H2O | −0.21 | 0.92 | −0.24 | 0.78 | −0.33 | 0.64 | −0.23 | 0.76 | |
*HCOO + H+ + e-→*HCOOH | −0.07 | 0.64 | −0.04 | 0.58 | −0.04 | 0.54 | −0.10 | 0.47 | |
为了评估CO2RR对各种产物的选择性,需要计算CO2加氢还原的第一步生成两个关键中间体的竞争反应的吉布斯自由能。微观动力学研究表明,*COOH是生成CO的关键中间体,而*HCOO是生成*HCOOH的关键中间体[6]。在此我们以Sb(101)表面为例,分析CO2RR应机理,如图1a所示(其余表面的自由能和活化能数据展示在表1中)。从热力学角度上来看,CO2生成*COOH和*HCOO反应自由能均大于0,在热力学上是非自发的,受电势的影响较大。因此这两个关键中间体的形成步骤均为电势限制步骤。在Sb(101)表面上,第一个电子转移步骤*CO2→*HCOO的ΔG为0.35 eV,比*CO2→*COOH的G = 0.80 eV小得多。这两个反应的能量来看,生成*HCOO的能量较低,所以甲酸路径是优势路径,在Sb(101)表面的还原产物为主要为甲酸,以及少量CO。根据表1反应自由能结果显示,其余三个表面上甲酸路径均占主导地位。
从动力学角度来揭示反应机理,基于Marcus微观动力学模型,计算了四种不同晶面上反应的活化能,如图1b所示。活化能越高,反应发生越困难。结果表明,反应生成*COOH的活化能垒远高于反应生成*HCOO,*COOH中间体在四个金属电极上都难以生成,而*CO2→*HCOO反应易于发生。因此,产物甲酸的选择性高于CO,是动力学控制的产物。其余表面的分析结果与Sb(101)相似,生成*COOH反应势垒均高于*HCOO,因此在Sb(110)、Bi(101)和Bi(110)表面上甲酸是动力学控制的主要产物。
实验上往往以催化活性来评判催化剂的好坏,也就是电流密度等指标。在理论计算中,我们可以通过反应速率来评估催化活性。为了更好的模拟与实验中电流-电位关系进行比较,图1c模拟了四种金属表面上*HCOO生成kHCOO的电位相关速率常数分布。速率常数定义为:
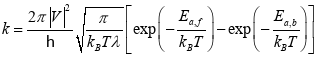 (5)
(5)
从速率常数随电压变化的曲线可以看出(图1c),对于Sb和Bi这两种P区金属来说,生成HCOOH的催化活性为Bi(101) > Bi(110) ≈ Sb(110) > Sb(101)。

图1.(a)电压为0 V时,CO2在Sb(101)表面上生成甲酸的势能曲线对比;(b)CO2生成*COOH和*HCOO的电势依赖反应势垒;(c)*HCOO生成的电位相关速率常数分布;(c)CO2RR和HER的自由能对比
析氢反应在CO2RR中是一个相当严重的副反应。副反应的程度对催化剂的选择和法拉第电流效率都会产生显著的影响。为了进一步理解解CO2还原反应与析氢反应的竞争关系,我们将析氢反应的第一步Volmer反应与CO2第一步加氢还原生成*HCOO的反应自由能进行对比,如图1d所示。相比于竞争性的*H,*HCOO表现出明显的能量优势,这一发现解释了Sb和Bi的四个金属表面上主要产物为甲酸的原因。
在这项工作中,我们基于Marcus电荷转移理论与速率常数理论相结合的微观动力学模型,研究了两种金属上电催化CO2还原产物的催化活性与选择性。确定了在四个金属表面上以生成HCOOH为主导的优势路径。通过理论模拟反应速率来判断催化活性,确定了Bi(101)表面较高的催化活性,计算结论与实验结果相吻合,催化活性顺序为:Bi(101) > Bi(110) ≈ Sb(110) > Sb(101)。
参考文献
(1)Sun, J. W.; Fu, H. Q.; Liu, P. F.; Chen, A.; Liu, P.; Yang, H. G.; Zhao, H. Advances and challenges in scalable carbon dioxide electrolysis. EES Catalysis 2023, 1, 934-949.
(2)Li, B.; Liu, L.; Yue, M.; Niu, Q.; Li, M.; Zhang, T.; Xie, W.; Wang, Q. Status and challenges for CO2 electroreduction to CH4: advanced catalysts and enhanced strategies. Green Chemistry 2024, 26, 103-121.
(3)Dattila, F.; Seemakurthi, R. R.; Zhou, Y.; López, N. Modeling Operando Electrochemical CO2 Reduction. Chem. Rev. 2022, 122, 11085-11130.
(4)Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A. V.; Wezendonk, T. A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO
2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804-9838.
(5)Wang, W.-H.; Himeda, Y.; Muckerman, J. T.; Manbeck, G. F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936-12973.
(6)Gao, S. T.; Xiang, S. Q.; Shi, J. L.; Zhang, W.; Zhao, L. B. Theoretical understanding of the electrochemical reaction barrier: a kinetic study of CO2 reduction reaction on copper electrodes. Phys. Chem. Chem. Phys. 2020, 22, 9607-9615.

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网 琼ICP备2021005105号



